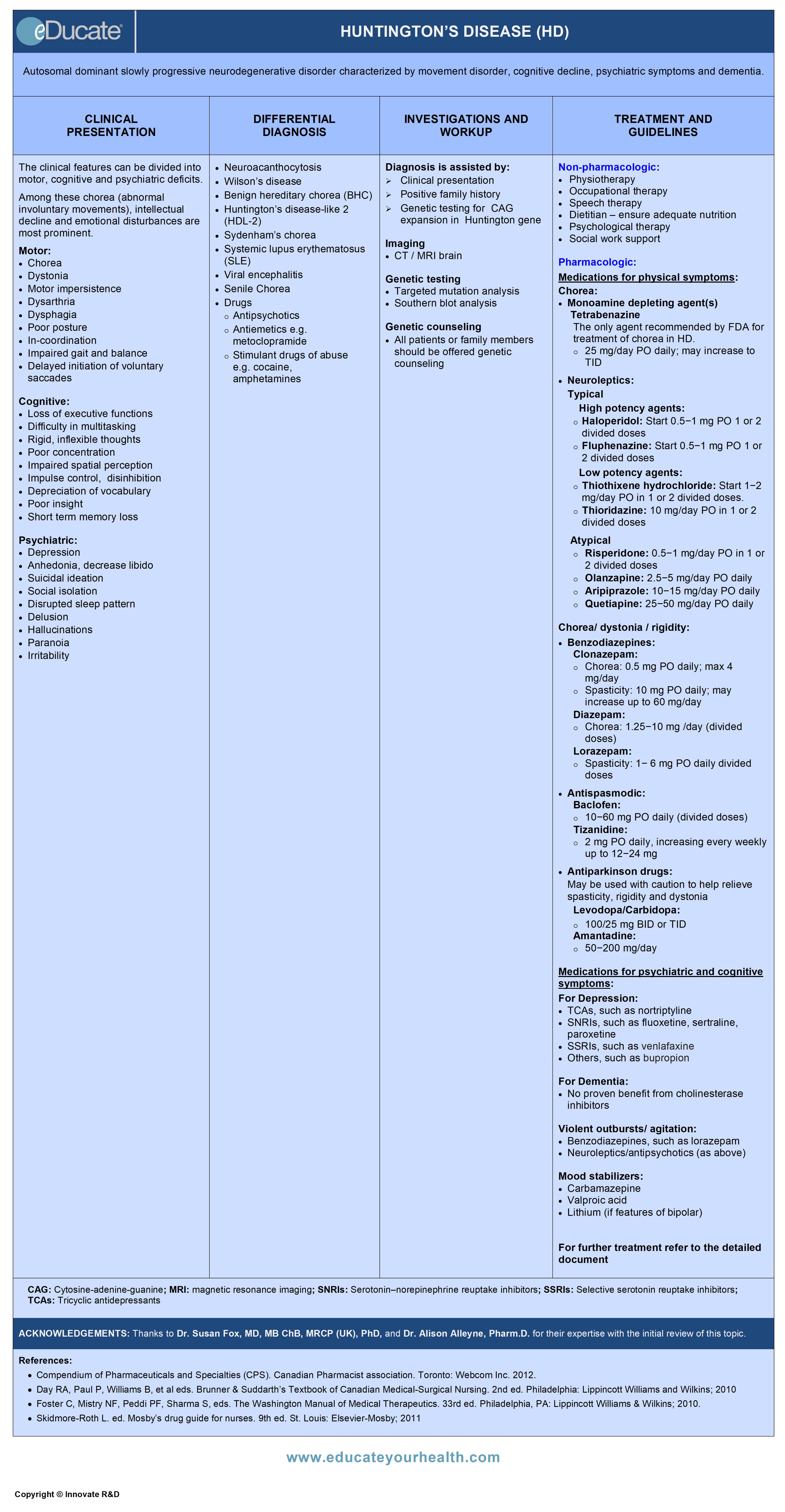
Huntington’s Disease (HD)
ACKNOWLEDGEMENTS:
Thanks to Dr. Susan Fox, MD, MB ChB, MRCP (UK), Ph.D., Associate Professor of Neurology, University Health Network, Toronto Western Hospital, University of Toronto, Canada and Dr. Alison Alleyne, Pharm.D., Coordinator, Clinical Pharmacy Services, Clinical Pharmacy Specialist, Neurosciences, Lower Mainland Pharmacy Services, Surrey, BC Canada for their expertise with the initial review of this topic.
Definition
Autosomal dominant progressive neurodegenerative disorder characterized by movement disorders, psychiatric symptoms and cognitive decline including dementia.
Etiology
Genetic disorder inherited in an autosomal dominant (AD) manner due to expansion of the cytosine-adenine-guanine (CAG) trinucleotide repeats in the huntingtin (HTT) gene (or HD or IT15 gene) and is located on chromosome 4.
Age of onset of the disease correlates with number of expanded repeats in the HTT i.e. at earlier age occurs with greater number of repeats.
- Children of an affected parent with Huntington’s disease have ~50% risk of developing the condition
- Abnormal expansion of the CAG triplet repeat within the HTT gene results in abnormal processing of the corresponding Huntingtin protein, which gradually leads to neuronal damage within the basal ganglia
- Large CAG expansions produce more widespread injury in the neurons
Epidemiology
- Most often between 40 and 50 years (mean age 45 years)
- 25% present after the age of 50 years
- 10% after the age of 60 years
Pathophysiology
- Under normal conditions, Huntingtin protein is present throughout the brain, and within a large number of tissues throughout the body
- Mutant Huntingtin gene with CAG expansion leads to transcription and expression of abnormal Huntingtin protein
- Selective neuronal loss within the caudate and putamen underlies the development of Huntington’s symptoms
- Thus despite widespread presence of abnormal huntingtin protein, selective neuronal vulnerability results in a specific pattern of cell loss and the clinical features of the disease
- The abnormal huntingtin protein is cleaved into smaller, toxic fragments that bind together and accumulate within neurons, disrupting the normal functions of these cells, including impaired axonal transport and synaptic transmission. Other potential mechanisms of neuronal cells death include:
- Increased oxidative stress
- Mitochondrial dysfunction
- Activation of proteases → leading to proteolysis
- Apoptosis
Clinical Presentation
The clinical features can be divided into motor, cognitive and psychiatric deficits. Among these chorea (abnormal involuntary movements), intellectual decline and emotional disturbances are most prominent.
{kind=link}
The presentation is divided into early, middle and late stage.
{kind=link}
Differential Diagnosis
Hereditary:
- Neuroacanthocytosis:
- Onset typically in 4th-5th decades
- Childhood and elderly cases can occur
- Chorea often milder than HD
- Other differentiating clinical features include: Personality changes, tics, obsessive-compulsive disorder (OCD), neuropathy, seizures. Test – blood smear for acanthocytes; elevated creatine kinase(CK), liver enzymes
- Benign hereditary chorea (BHC): Autosomal dominant, non-progressive movement disorder, with onset in infancy or early childhood. No cognitive or psychiatric changes
- Wilson’s disease: Autosomal recessive disorder with abnormal copper metabolism leading to accumulation of copper in liver and other organs including brain, kidneys, and eyes. Manifests with movement disorders and psychiatric/behavioral abnormalities. Dystonia and tremor are more common in Wilson’s disease compared to HD. Investigations include 24h urinary copper collection, serum ceruloplasmin and ophthalmological evaluation for the presence of Kayser-Fleischer rings
- Dentatorubropallidoluysian atrophy (DRPLA)
- Autosomal dominant (AD), Japanese ancestry
- Clinically mixed chorea, myoclonus, dystonia, cognitive decline, ataxia, tremor
- Spinocerebellar ataxias (SCA)1,2,3, and 17
- Cerebellar ataxia plus psychiatric symptoms, and chorea
- Neuroferritinopathy
- AD inheritance; mutation of light chain ferritin, low serum ferritin
- MRI – shows iron deposition
- Huntington’s disease-like 2 (HDL-2)
- African ancestry
- Autosomal dominant inheritance caused by a CTG/ repeat expansion in the junctophilin gene
- Chorea and dementia 3-4th decade
Autoimmune:
- Sydenham’s chorea: Autoimmune etiology secondary to exposure to group A beta-hemolytic streptococcus
- Systemic lupus erythematosus (SLE): Associated with presence of prothrombotic antiphospholipid antibodies (lupus anticoagulant)
Infectious:
- Viral encephalitis, HIV, TB, syphilis, Lyme disease may all cause chorea
Drugs:
- Antiemetics e.g. metoclopramide can also cause orofacial and limb dyskinesia
- Stimulant drugs of abuse e.g. cocaine, amphetamines may induce personality change and chorea
- Antipsychotics
- Tardive dyskinesia: Classically orofacial stereotypic movements, as well as limb chorea. Differentiating from chorea in HD
- Tardive dyskinesia (TD) is usually suppressible e.g. when patient eats in TD movements are less compared to HD where food may fall out of the mouth
Metabolic/endocrine disorders:
- Chorea gravidarum
- Hyperthyroidism
- Hyperosmolar hyperglycemic non-ketotic syndrome may have chorea-ballism as an accompanying feature, likely due to hypoperfusion of the striatum
- Acquired hepatolenticular degeneration; orofacial dyskinesia can occur in chronic liver disease due to manganese deposition in the striatum
- Idiopathic calcification in the basal ganglia (Fahr’s disease); get a CT brain scan
Vascular:
- Ischemic or hemorrhagic lesion of the subthalamic nucleus may result in contralateral hemi-ballisms and hemi-chorea. Onset is often abrupt, may persist but often diminishes over time
- Cerebral hypoperfusion that might occur with prolonged cardiac bypass (on-pump time) or circulatory arrest may result in a post-pump syndrome that includes choreoathetosis
Senile Chorea:
- Late-onset, non-familial chorea, often involving the face, tongue, and mouth. Various metabolic conditions have been implicated including hypocalcemia and antiphospholipid antibody syndrome
Investigation and Workup
Diagnosis is assisted by:
➣ Clinical presentation
➣ Positive family history
➣ Genetic testing for CAG expansion in the huntingtin gene
Imaging:
- CT/ MRI brain
Genetic testing:
- Targeted mutation analysis:
- PCR-based methods detect alleles up to about 115 CAG repeats
- Southern blot analysis:
- Identification of large expansions which may fail to amplify well by PCR analysis associated with juvenile-onset HD
- Confirmation of apparent homozygous genotypes obtained by PCR analysis

Genetic counselling:
All patients or family members should be offered genetic counselling, but not all patients may want or need genetic testing.
Issues to be addressed are as follows:
Inheritance pattern:
- Each child of an affected individual has a 50% chance of inheriting the abnormal huntingtin gene
- Inheriting a normal huntingtin gene from the unaffected parent does not prevent or counteract the disease-causing effects of the abnormal gene
- Greater expansion with paternal inheritance
Absent family history of HD:
- ~2-5% of all cases develop HD without knowing that they were at risk
- Parent carrying a gene did not live long enough to manifest the symptoms)
- Non-paternity
Usefulness of genetic testing:
- Diagnostic or confirmatory testing – (if the clinical suspicion is strong then it is the only diagnostic test needed)
- Predictive or presymptomatic testing** (recommended in individuals with no symptoms, but wishes to know if he carries the expanded gene). Current implications are that no cure or treatment to slow down disease progression currently exists
- ** May pose potential psychosocial risks and hence often requires different clinical approach with appropriate counselling.
- Prenatal testing: Prenatal testing for HD is possible, and should be performed in conjunction with detailed genetic counselling
Treatment
Treatment for HD is largely symptomatic and highly individualized to help control physical and mental deficits.
Treatment strategies vary over time and from individual to individual, even within a family.
Always consider non-pharmacological interventions first.
Goals of treatment:
- Counsel patient and family to improve awareness regarding prognosis, and understanding of the disease course
- Symptomatic improvement
- Maintain functionality for as long as possible
TREATMENT STRATEGY:
- Non-pharmacological
- Pharmacological
Non-pharmacological therapy:
Can be used to assist physical disabilities and mental conditions and includes:
- Physiotherapy: Improve/maintain posture, strength, coordination, balance, gait
- Occupational therapy: Assess for safety and need for assistive devices. Improve ambulation, avoid falls, and improve nutrition (eating)
- Speech therapy: Improve/maintain speech and swallowing
- Dietitian: To ensure adequate nutrition
- Psychological therapy: Counsel patient and family members; assist in coping with skills
- Social work support: Help family prepare for deteriorating clinical course
- Assist with arrangements for (full time) supervision as required
- Suggest and assist with identification of alternate living arrangements (assisted living, nursing home) if required
Pharmacological therapy:
To date, the only FDA recommended drug for use in HD is tetrabenazine to treat chorea. There is currently a lack of evidence-based medicine recommendations due to few class I, randomized controlled trials (RCTs) of agents for any of the symptoms and thus no treatment guidelines exist.
The current choice of agents for any of the motor, psychiatric and cognitive symptoms relies on expert opinion, physicians’ personal choice and knowledge of an agent balanced against potential side-effect profile.
For convenience; therapy is documented according to symptoms during the disease course.
Medications for physical symptoms:
Chorea: A prominent movement disorder of HD which changes over time, eventually peaks and then begins to decline. Treatment is initiated when chorea begins to interfere with voluntary activities like writing, cooking, eating or causes injury due to fall or accidents. It is worsened by stress, anxiety, depression and lack of sleep. Not all patients require treatment and the need has to be balanced against side-effects.
(a) Monoamine depleting agents:
- Tetrabenazine: 25 mg/day PO daily; may increase to TID
(b) Dopamine D2 antagonists:
May treat both chorea and some psychiatric symptoms.
Typical antipsychotics:
- Haloperidol: Start at low dose 0.5-1 mg PO in 1 or 2 divided doses, with gradual increase
- Fluphenazine: Start at low dose 0.5-1 mg PO in 1 or 2 divided doses, with gradual increase
- Thiothixene hydrochloride: Start with1-2 mg/day PO in 1 or 2 divided doses
- Thioridazine: 10 mg/day PO in 1 or 2 divided doses
Atypical antipsychotics:
- Risperidone: 0.5-1 mg/day PO in 1 or 2 divided doses
- Olanzapine: 2.5-5 mg/day PO daily
- Aripiprazole: 10-15 mg/day PO daily
- Quetiapine: 25-50 mg/day PO daily
(c) Amantadine – may be helpful for mild chorea
(d) Benzodiazepines – see below
Dystonia, rigidity, Parkinsonism, and spasticity:
With disease progression, the clinical phenotype changes from hyperkinetic to hypokinetic. Thus dystonia may become more evident with more sustained muscle contractions. Rigidity and parkinsonism are also evident with advanced disease. In Juvenile onset (due to the larger number of CAG repeats); this hypokinetic phenotype is often the presenting feature with parkinsonism, plus spasticity and cerebellar ataxia.
(a) Benzodiazepines:
- Clonazepam:
- Dose in chorea: 0.5-5 mg/day PO
- Dose in spasticity and stiffness: 10 mg/day PO increasing up to 60 mg/day in divided doses
- Diazepam:
- For Chorea: 1.25 mg/day in divided doses up to 10 g/day
- Lorazepam: 1-6 mg PO daily in divided doses
(b) Antispasmodic:
- Baclofen: 10-60 mg PO daily in divided doses
(c) Alpha 2-adrenergic agonist:
- Tizanidine: 2 mg PO daily, increasing every week up to 12-24 mg, for spasticity with or without baclofen
(d) Antiparkinson drugs:
May also be used (cautiously) to relieve spasticity, rigidity, dystonia and includes
- Levodopa/carbidopa: 100/25 mg 2-3 times per day
(e) Botox: For focal dystonia
Medications for psychiatric and cognitive symptoms:
Depression:
(a) Tricyclic antidepressants (TCA):
- Nortriptyline: 10-25 mg PO daily, up to 150-200 mg daily
(b) Selective serotonin reuptake inhibitors (SSRI):
- Fluoxetine: 10-20 mg PO daily up to 60-80 mg
- Sertraline: 25-50 mg PO daily, up to the max of 200 mg
- Paroxetine: 10-20 mg PO daily, up to 40-60 mg
(c) Selective norepinephrine reuptake inhibitors (SNRI):
- Venlafaxine: Start 25-37.5 mg PO; may increase up to 225 mg/day in divided doses or once daily (XR)
(d) Others:
- Buproprion: 100-200 mg PO daily in divided doses, max dose 300-450 mg
Dementia:
No specific treatment. Cholinesterase inhibitors (donepezil, rivastigmine, and galantamine) have been tried, but no proven benefit.
Violent outbursts/agitation:
(a) Benzodiazepines:
Short-acting drugs such as lorazepam may be another good choice, for the short-term management of agitation
- Lorazepam: 1-2 mg/day PO at bedtime or 2-3 times a day
(b) Neuroleptics/antipsychotics (as above)
Mood stabilizers/mania:
- Valproic acid: 125-250 mg PO BID; increase dose gradually to an effective level or reach a serum concentration of 50-120 mcg/mL or in SI units (400-700 Umol/L); Max. 500-2000 mg PO BID
- Carbamazepine: 100-200 mg PO daily increase gradually by 100 mg/day as tolerated to ~1200-1600 mg/day in 2-3 divided doses
- Lithium (particularly if features of bipolar): Start 900-2100 mg (15-20 mg/kg/day) in 3 divided doses – adjust dose to target levels range from 0.8-1.2 mmol/L, as needed and tolerated
[/cq_vc_tab_item][cq_vc_tab_item tabtitle=”Medication Dose”]MEDICATIONS:
{kind=link}
- ➣ Fluphenazine
- ➣ Haloperidol
- ➣ Thiothixene
Potential Mechanism(s):
- Blocks postsynaptic dopaminergic receptors in the brain
- Depresses the release of hypothalamic and hypophyseal hormones
- Also have weak anticholinergic activity and antiemetic effect
Doses:
Fluphenazine
Psychosis:
- Start 0.5-2.5 mg PO and increase to 2.5-10 mg/day in divided dose at 6-8 hrs interval; usual maintenance dose 1-5 mg once daily; Max. 20-30 mg/day
Chorea:
- Start 0.5-1 mg/day PO; Max. 6-8 mg/day
Haloperidol
Psychosis:
- Start 0.5-2.5 mg PO and increase to 0.5-5 mg PO BID-TID; maximum usual dose: 20-30 mg/day
Chorea:
- Start 0.5-1 mg/day PO; Max. 6-8 mg/day
Thioridazine
Psychosis:
- 10 mg/day PO in 1 or 2 divided doses
Chorea:
- Start 10 mg/day PO in divided doses; max dose 100 mg/day
Thiothixene
Mild-to-moderate psychosis:
- Start: 5 mg PO BID, increase gradually up to 20-30 mg/day as required: Max 60 mg/day
Rapid tranquilization of the agitated patient
(administered every 30-60 minutes):
- 5-10 mg PO; average total dose for tranquilization: 15-30 mg
Chorea: 1-2 mg/day PO; max dose up to 10-20 mg/day
{kind=link}
- ➣ Aripiprazole
- ➣ Olanzapine
- ➣ Risperidone
- ➣ Quetiapine
Potential Mechanism(s):
- Exact mechanism(s) of action is unknown
- Potent antagonist of serotonin 5-HT2a, dopamine D2, receptors
- Potentially blocks dopamine transmission and treat chorea and psychiatric symptoms
Doses:
Aripiprazole
Psychosis:
- 10-15 mg PO once daily; if needed, may increase dose gradually to maximum of 30 mg PO once daily at minimum of 2 weeks of interval
Olanzapine
Psychosis:
- 2.5-5 mg PO once daily and increase as tolerated 10 mg PO once daily within 5-7 days as required; may increase by 5 mg/day at weekly intervals. Usual maintenance dose 10-20 mg once daily; Max. 15-20 mg/day
- Usual dose in elderly is 1.25-7.5 mg
Risperidone
Psychosis/chorea:
- May begin with 0.5-1 mg PO once or twice daily. Increase gradually by 1-2 mg/day on a weekly basis to target dose of 4-8 6 mg/day
- For psychosis may increase as tolerated to max. dose of 4-6 mg/day
Quetiapine
Psychosis:
- Start 25 mg PO BID; followed by increments of 25-50 mg daily divided over 2-3 doses, as tolerated to a target dose of 300-400 mg/day in 2-3 divided doses within 4 days (if needed). May increase further by 25-50 mg divided BID every 2-3 days if required; usual maintenance range: 300-800 mg/day
- Hepatic impairment: Initially 25 mg/day, increase dose by 25-50 mg/day to an effective dose, based on clinical response and tolerability to patient
{kind=link}
- ➣ Tetrabenazine
Mechanism:
- Reversibly inhibits uptake of monoamine neurotransmitters in presynaptic vesicles
- Inhibits presynaptic dopamine release and also blocks CNS dopamine receptors
- Also depletes monoamine stores from nerve terminals
Doses:
Tetrabenazine
Hyperkinetic movement disorders:
- Start 12.5 mg PO BID or TID; may be increased by 12.5 mg/day every 3-5 days slowly to maximal tolerated and effective dose, if needed; usual maximum tolerated single dose 25 mg PO in poor metabolizers
- Daily dosage >100 mg/day are not recommended
- However, some have reported that doses up to 200 mg/day have been used on rare occasions
{kind=link}
- ➣ Clonazepam
- ➣ Diazepam
- ➣ Lorazepam
Mechanism:
- Benzodiazepines bind to the gamma sub-unit of the GABA receptor and enhance the inhibitory effect of GABA
- Increase the frequency of channel opening events, leads to
- Increase in chloride ion conductance and inhibition of the action potential
- All benzodiazepines exert five major effects: (i) Anxiolytic (ii) Hypnotic (iii) Muscle relaxant (iv) Anticonvulsant (v) Amnesia (transient loss of memory)
Doses:
Clonazepam
Chorea:
- Start 0.5 mg/day PO; may increase to 4 mg/day in divided doses as tolerated
Spasticity/ rigidity:
- 10 mg/day PO; may increase up to 60 mg/day as needed and tolerated
Diazepam
Chorea:
- 1.25 mg/day in divided doses up to 20 mg/day in divided doses
Lorazepam
Chorea:
- 1-6 mg PO daily in divided doses
Violent outbursts/ agitation:
- 1-2 mg/day PO at bedtime or 2-3 times a day
{kind=link}
- ➣ Baclofen
- ➣ Tizanidine
Mechanism:
Complete mechanism of action unknown.
Baclofen
- It inhibits both monosynaptic and polysynaptic spinal reflexes, possibly by hyperpolarization of afferent terminals (resultant relief of muscle spasticity)
Tizanidine
- It is chemically-related to clonidine and other α2-adrenergic agonists, which acts as a centrally acting muscle relaxant with anti-hypertensive properties
- Presumably, it reduces spasticity by increasing presynaptic inhibition of motor neurons
Dose:
Baclofen
Spasticity/ rigidity:
- 10 mg/day PO; may increase up to 60 mg/day as needed and tolerated
Tizanidine
- 2 mg PO daily at bed time, may increase weekly up to maximum of 12-24 mg/day in divided dose, ( used with or without baclofen)
{kind=link}
- ➣ Levodopa/ carbidopa
Mechanism:
- Act on dopamine receptors in striatum to modulate basal ganglia function
- Carbidopa and B benserazide inhibit peripheral breakdown of Levodopa (L-Dopa) by inhibiting decarboxylation, thereby increasing availability of L-Dopa to cross the blood-brain barrier (BBB)
- Within the basal ganglia, L-Dopa is then converted to dopamine
Dose:
Levodopa/ carbidopa
- 25/100 mg PO two to three times per day
{kind=link}
- ➣ Amantadine
Mechanism:
Possibly enhances dopamine action by:
- Blocking the reuptake of dopamine into presynaptic neurons
- Increase dopamine release from presynaptic fibers
- Reduces levodopa-induced dyskinesia by antagonism of NMDA glutamate receptors
Dose:
- 50-200 mg per day PO
Tricyclic antidepressants (TCAs)
{kind=link}
- ➣ Nortriptyline
Use: Depression
Mechanism:
- Inhibits the presynaptic reuptake of neurotransmitters → increases synaptic serotonin and/or norepinephrine → potentiates its effect
- Also has significant anticholinergic properties
Dose:
Nortriptyline
- 10-25 mg PO daily; may increase gradually to 150-200 mg if needed
Selective serotonin reuptake inhibitors
{kind=link}
- ➣ Fluoxetine
- ➣ Paroxetine
- ➣ Sertraline
Use: Depression
Mechanism:
- Inhibits the presynaptic reuptake of neurotransmitters, in turn, increases synaptic serotonin concentration and potentiates its effect
- Little effect on the reuptake of norepinephrine or dopamine
- Does not significantly bind to alpha-adrenergic, histamine, or cholinergic receptors
Dose:
Fluoxetine
- Start 10 mg PO; may increase to 20 mg after at least 1 week; Max. 60-80 mg
Paroxetine
- 10 mg PO; may increase to 20 mg after at least 1 week, if well tolerated; Max. 40-60 mg
Sertraline
- 25-50 mg PO; may increase to 50-100 mg after at least 1 week; Max. 200 mg
Serotonin/Norepinephrine reuptake inhibitor (SNRI)
{kind=link}
- ➣ Venlafaxine
- ➣ Duloxetine
- ➣ Nefazodone
Mechanism:
Exact mechanism of action is unknown.
- A potent serotonin and norepinephrine reuptake inhibitor
- Weakly inhibits dopamine reuptake
- Duloxetine has similar affinity to NE and 5-HT receptors (Ki ratio 9) whereas venlafaxine has a relatively higher binding affinity to norepinephrine
Dose:
Venlafaxine
- Start 25-37.5 mg PO; may increase up to 225 mg/day
Duloxetine
- Start 30 mg PO daily; may increase 30 mg/week up to 120 mg/day
Nefazodone
- 50-100 mg 450-600 mg
Antidepressant, Dopamine reuptake inhibitor
{kind=link}
- ➣ Bupropion
Mechanism:
Exact mechanism of action is unknown.
- Binds selectively to the dopamine transporter, but its behavioural effects have often been attributed to its inhibition of norepinephrine reuptake
- Often acts as nicotinic acetylcholine receptor antagonist
Dose:
Bupropion
- 100-200 mg PO daily in divide doses, max. dose 300-450 mg
{kind=link}
- ➣ Valproic acid
- ➣ Carbamazepine
- ➣ Lithium (particularly if features of bipolar)
Mechanism:
Carbamazepine
- Blocks voltage-gated sodium channels and may potentiate GABA receptors; decreases synaptic transmission within CNS
Valproic acid (valproate)
- Blocks voltage-gated sodium channels and T-type calcium channels. Also affects GABA resulting in decreased synaptic transmission within CNS
Lithium
- Alters sodium transport in nerve and muscle cells and Inhibit the intracellular formation of cyclic AMP; enhances serotonin transmission by reducing the activity of post-synaptic serotonin or 5-HT receptors
Use: Acute manias/ mood stabilizers
Dose:
Carbamazepine
- Start 100-200 mg/day; increase dose gradually by 100 mg/day as tolerated to an effective level or therapeutic level of 5-12 mcg/ml; Max. 1200-1600 mg/day
Valproic acid (valproate)
- Start 125-250 mg PO BID; increase dose gradually to effective level or reach a blood level of 50-150 mcg/ml; Max. 1000-2000 mg PO divided BID or TID
Lithium
- Start 900-2100 mg (15-20 mg/kg/day) in 3 divided doses – adjust dose to target levels range from 0.8-1.2 mmol/L, as needed and tolerated
- For maintenance therapy, adjust the dose to maintain serum lithium concentrations between 0.6 and 1 mmol/L; once the patient is stabilized than change maintenance dosage to once daily regimen
Renal impairment:
- CrCl 10-50 mL/minute: Administer 50% to 75% of normal dose
- CrCl <10 mL/minute: Administer 25% to 50% of normal dose
[/cq_vc_tab_item][/cq_vc_tabs]
{kind=link}
{kind=link}
Physician Resources
1. Tips for Patient Care
General:
- Educate patients, caregivers, and family about HD including anticipated clinical course and treatment
- If dysarthria is present, caregivers should allow enough time for the patient to communicate
- If dysphagia is present patients are encouraged to concentrate on chewing and swallowing; avoid talking while eating
- Ensure the patient has adequate nutrition
- Patients should be encouraged to openly express their concerns and feelings as this might help reveal cognitive/psychiatric concerns such as suicidal ideation
Activities:
- Encourage patient to participate fully in various physical activities, including activities of daily living, as long as possible and if safe enough to do so, but avoid overwork and fatigue.
- Encourage patient to participate in activities of daily living, if it is safe enough to do so. Advise to avoid fatigue and overwork
- Patients should get involved in learning relaxation techniques
- If the patient is obese weight loss may decrease stress on weight-bearing joints, and help with mobility
Counseling:
- Genetic counseling/testing may be considered if there is a family history of Huntington’s disease; this however remain a personal decision and varies from patient to patient. A skilled genetic counsellor would be helpful
- Other areas that may require counselling includes:
- Requirement for assistive devices and mobility aids
- Feeding tubes for patients unable to swallow
- Placement/nursing home
- Power of attorney for handling legal affairs
Monitoring:
Patients should be monitored for:
- Fall risk
- Behavioural changes
- Response to therapy, and side effects of any treatments
- Suicidal thoughts
Medications:
- Treatment requires integrated, multidisciplinary approach
- Treatment should be individually tailored
- Medications should begin with low doses, whenever possible
- Currently, no treatment can alter, slow, or reverse the progression of HD
- Treatment is to palliate the symptoms; causing distress
- Consider concurrent risk factors and disease states with the prescribed therapy
- Monitor all patients for notable behaviour changes, such as suicidal thoughts
- Discuss potential adverse events/side effects of medications
- Evaluate cost, affordability and insurance coverage for patients
Social and stress factors:
- Chorea is exacerbated by stress, anxiety, or depression
- Patient and family members are often confronted with the social challenges surrounding the physical disability as well as the psychiatric aspects of impulse control, disinhibition, and outbursts
Tips:
- Symptoms may vary from individual to individual, even within a family
- Chorea is the most evident feature of HD and may diminish over time
- Chorea does not always need to be treated as side-effects of currently available drugs may out way benefit
- The most common specific psychiatric disorder in HD is depression
- As dysphagia develops, a soft diet with liquid supplements may be needed. However, complications surrounding dysphagia is the most common cause of death in people with late-stage Huntington’s disease
- Suicide is the second commonest cause of death in HD
- Apathy is also common in HD and can be hard to distinguish from depression
Expected outcome:
- There is no cure for Huntington’s disease
- Progressive impairment
- Life expectancy is ~15 to 20 years from diagnosis
2. Scales and Table
References
Core Resources:
- Compendium of Pharmaceuticals and Specialties (CPS). Canadian Pharmacist Association. Toronto: Webcom Inc. 2012
- Day RA, Paul P, Williams B, et al (eds). Brunner & Suddarth’s Textbook of Canadian Medical-Surgical Nursing. 2nd ed. Philadelphia: Lippincott Williams and Wilkins; 2010
- Foster C, Mistry NF, Peddi PF, Sharma S, eds. The Washington Manual of Medical Therapeutics. 33rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010
- Gray J, ed. Therapeutic Choices. Canadian Pharmacists Association. 6th ed. Toronto: Webcom Inc. 2011
- Katzung BG, Masters SB, Trevor AJ, eds. Basic and Clinical Pharmacology. 11th ed. New York: McGraw-Hill; 2009
- Longo D, Fauci A, Kasper D, et al (eds). Harrison’s Principles of Internal Medicine. 18thed. New York: McGraw-Hill; 2011
- McPhee SJ, Papadakis MA, eds. Current Medical Diagnosis & Treatment. 49th ed. New York: McGraw-Hill; 2010
- Pagana KD, Pagana TJ eds. Mosby’s Diagnostic and Laboratory Test Reference. 9th ed. St. Louis: Elsevier-Mosby; 2009
- Skidmore-Roth L. ed. Mosby’s drug guide for nurses. 9th ed. St. Louis: Elsevier-Mosby; 2011
- Skidmore-Roth L, ed. Mosby’s nursing drug reference. 24th ed. St. Louis: Elsevier-Mosby; 2011
- Online Resources:
- http://bjp.rcpsych.org/content/149/6/682
- http://www.ncbi.nlm.nih.gov/books/NBK1305/
- http://www.hdfoundation.org/html/testwfn.php
- http://www.hdac.org/features/article.php?p_articleNumber=519
- http://www.huntingtonsociety.ca/english/content/?page=91
- http://www.hdac.org/features/article.php?p_articleNumber=519
- http://ghr.nlm.nih.gov/gene/HTT
- www.wemove.org
Online Pharmacological Resources:
- e-Therapeutics
- Lexicomp
- RxList
- Epocrates
Journals/Clinical Trials:
- Bonelli RM, Wenning GK. Pharmacological management of Huntington’s disease: an evidence-based review. Curr Pharm Des 2006; 12:2701
- de Yebenes JG, Landwehrmeyer B, Squitieri F, Reilmann R, Rosser A, Barker RA, Saft C, Magnet MK, Sword A, Rembratt A, Tedroff J; MermaiHD study investigators.Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011 Dec; 10(12):1049-1057
- Duff K, Paulsen J, Mills J, et al. Mild cognitive impairment in prediagnosed Huntington disease. Neurology 2010; 75:500
- Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci 2008; 27:2803
- Huntington Study Group (HSG) A Phase III, Randomized, Double-Blind, Placebo-Controlled, Safety and Efficacy Study of Dimebon in Patients with Mild-To-Moderate Huntington Disease (http://www.hdsa.org/research/clinical-trials/ongoing-clinical-trials/dimebontrial.html)
- Kegelmeyer D, Fritz N, Kostyk S, Kloos A, et al. The effect of video game-based exercise on dynamic balance and mobility in individuals with Huntington’s disease. J Neurol Neurosurg Psychiatry 2010;81:A40 doi:10.1136/jnnp.2010.222661.4
- Leigh RJ, Newman SA, Folstein SE, et al. Abnormal ocular motor control in Huntington’s disease. Neurology 1983; 33:1268
- Mestre T, Ferreira J, Coelho MM, et al. Therapeutic interventions for symptomatic treatment in Huntington’s disease. Cochrane Database Syst Rev 2009;CD006456
- Ondo WG, Tintner R, Thomas M, Jankovic J. Tetrabenazine treatment for Huntington’s disease-associated chorea. Clin Neuropharmacol 2002; 25:300
- Potter NT, Spector EB, Prior TW. Technical standards and guidelines for Huntington disease testing. Genet Med 2004:6:61-65
- Pringsheim T, Wiltshire K, Day L, et al. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord 2012; 27:1083
- Seneca S, Fagnart D, Keymolen K, et al. Early onset Huntington disease: a neuronal degeneration syndrome. Eur J Pediatr 2004; 163:717
- Walker FO. Huntington’s disease. Lancet 2007; 369:218
- Warby SC, Graham RK, and Hayden MR. Huntington Disease. NCBI Bookshelf. Initial Posting: October 23, 1998; Last Update: April 22, 2010